RNA-stability
unawaz1996
2023-05-05
Last updated: 2023-05-12
Checks: 6 1
Knit directory: NMD-analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230314) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /home/neuro/Documents/NMD_analysis/Analysis/NMD-analysis/data/LTK_Sample Metafile_V3.txt | data/LTK_Sample Metafile_V3.txt |
| /home/neuro/Documents/NMD_analysis/Analysis/NMD-analysis/data/data2.txt | data/data2.txt |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version bff9b8f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/Differential-transcript-usage.nb.html
Ignored: analysis/Enichment-analysis-fgsea.nb.html
Ignored: analysis/Enichment-analysis-goseq.nb.html
Untracked files:
Untracked: PCA.png
Untracked: PCA_plot.pdf
Untracked: PCA_transcript.png
Untracked: analysis/Differential-transcript-usage.Rmd
Untracked: analysis/UPF3B_KD.Rmd
Untracked: analysis/transcript-preprocessing.Rmd
Untracked: code/eisaR.R
Untracked: code/external_code/
Untracked: data/LTK_Sample Metafile_V3.txt
Untracked: data/Mus_musculus.GRCm39.105__nifs.tsv
Untracked: data/data.txt
Untracked: data/data2.txt
Untracked: data/fastqc/
Untracked: data/nif_output/
Untracked: data/samples.txt
Untracked: output/DEG-limma-results.Rda
Untracked: output/DEG-list.Rda
Untracked: output/DEG/
Untracked: output/EISA/
Untracked: output/ISAR/
Untracked: output/QC/
Untracked: output/Transcript/
Untracked: output/isoformSwitchAnalyzeR_isoform_AA_complete.fasta
Untracked: output/isoformSwitchAnalyzeR_isoform_AA_subset_1_of_3.fasta
Untracked: output/isoformSwitchAnalyzeR_isoform_AA_subset_2_of_3.fasta
Untracked: output/isoformSwitchAnalyzeR_isoform_AA_subset_3_of_3.fasta
Untracked: output/isoformSwitchAnalyzeR_isoform_nt.fasta
Untracked: output/limma-matrices.Rda
Untracked: tmp/
Unstaged changes:
Modified: analysis/_site.yml
Modified: code/functions.R
Modified: code/libraries.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/RNA-stability.Rmd) and
HTML (docs/RNA-stability.html) files. If you’ve configured
a remote Git repository (see ?wflow_git_remote), click on
the hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | bff9b8f | unawaz1996 | 2023-05-12 | Build site. |
| Rmd | 05f4d8c | unawaz1996 | 2023-05-12 | wflow_publish(c("analysis/index.Rmd", "analysis/DEG-analysis.Rmd", |
| html | 01ff011 | unawaz1996 | 2023-05-08 | Build site. |
| Rmd | f4e0d39 | unawaz1996 | 2023-05-08 | wflow_publish("analysis/RNA-stability.Rmd") |
Introduction
RNA-stability analysis
It has been suggested that, while exonic read counts in RNA-seq data correspond to steady-state mRNA abundance, changes in the abundance of intronic reads can be used to estimate the change in transcription rate. Through this concept, a change in exonic reads without a corresponding change in intronic reads is diagnostic of differential RNA stability, while concurrent changes in both exonic and intronic reads suggest altered transcription. DiffRAC is a framework that converts unspliced/spliced relationships into a generalized linear model whose parameters can then be inferred from sequencing count data.
Analysis
Initializing DiffRAC framework...
Estimating size factors and dispersions...
Optimizing the bias constant...
0.381966011250105 : 443084.805542457
0.618033988749895 : 451660.927947527
0.76393202250021 : 447436.811077347
0.606281593377457 : 451755.93420737
0.581622715887764 : 451809.600721294
0.587245246809309 : 451815.294125421
0.587578588893278 : 451815.288818795
0.586911904725339 : 451815.259613898
0.587245246809309 : 451815.294125421
The bias constant is 0.587245246809309
Re-estimating dispersion...
Fitting model parameters...Differentially stabilised genes
UPF3B
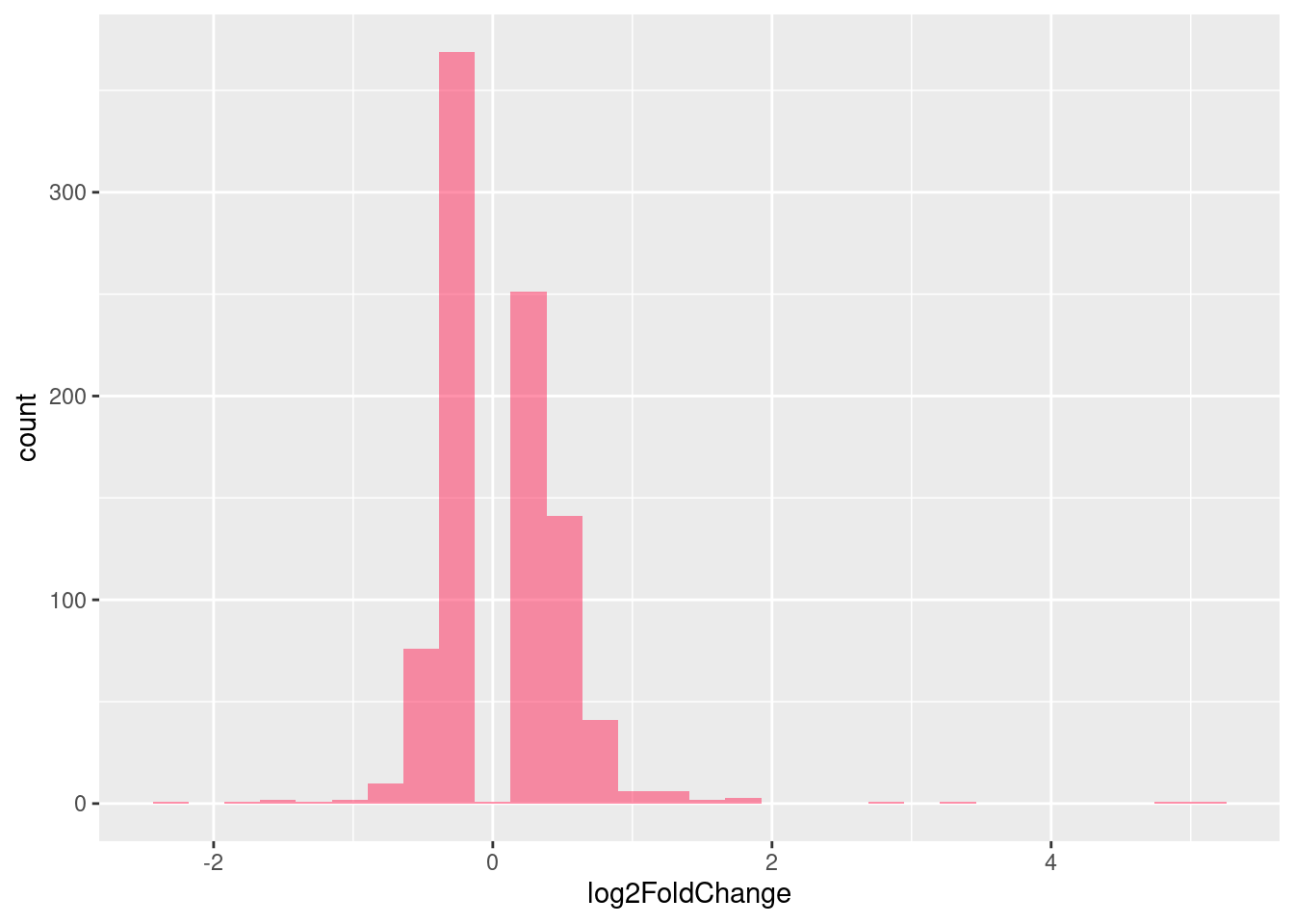
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
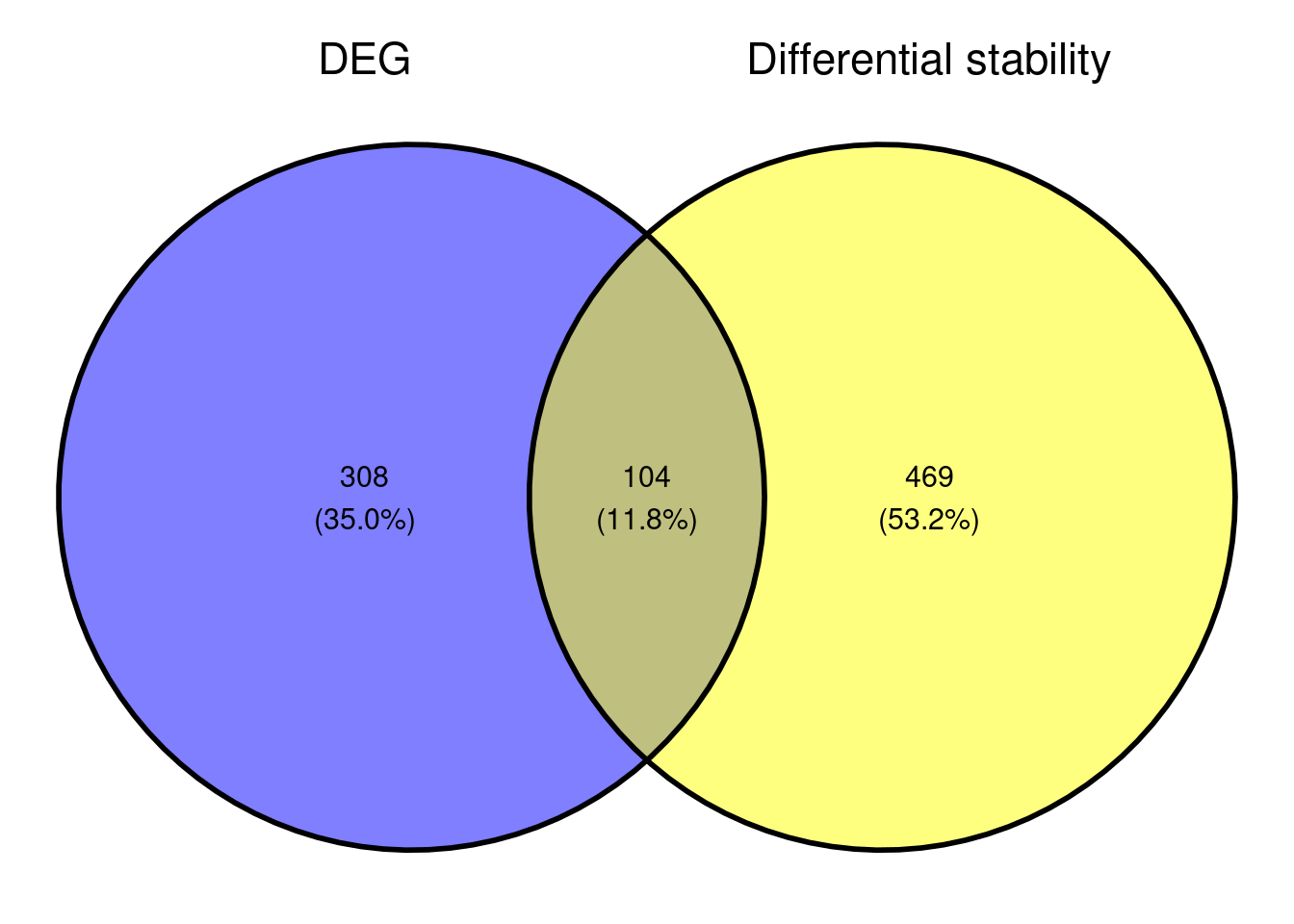
Comparison with DEG results
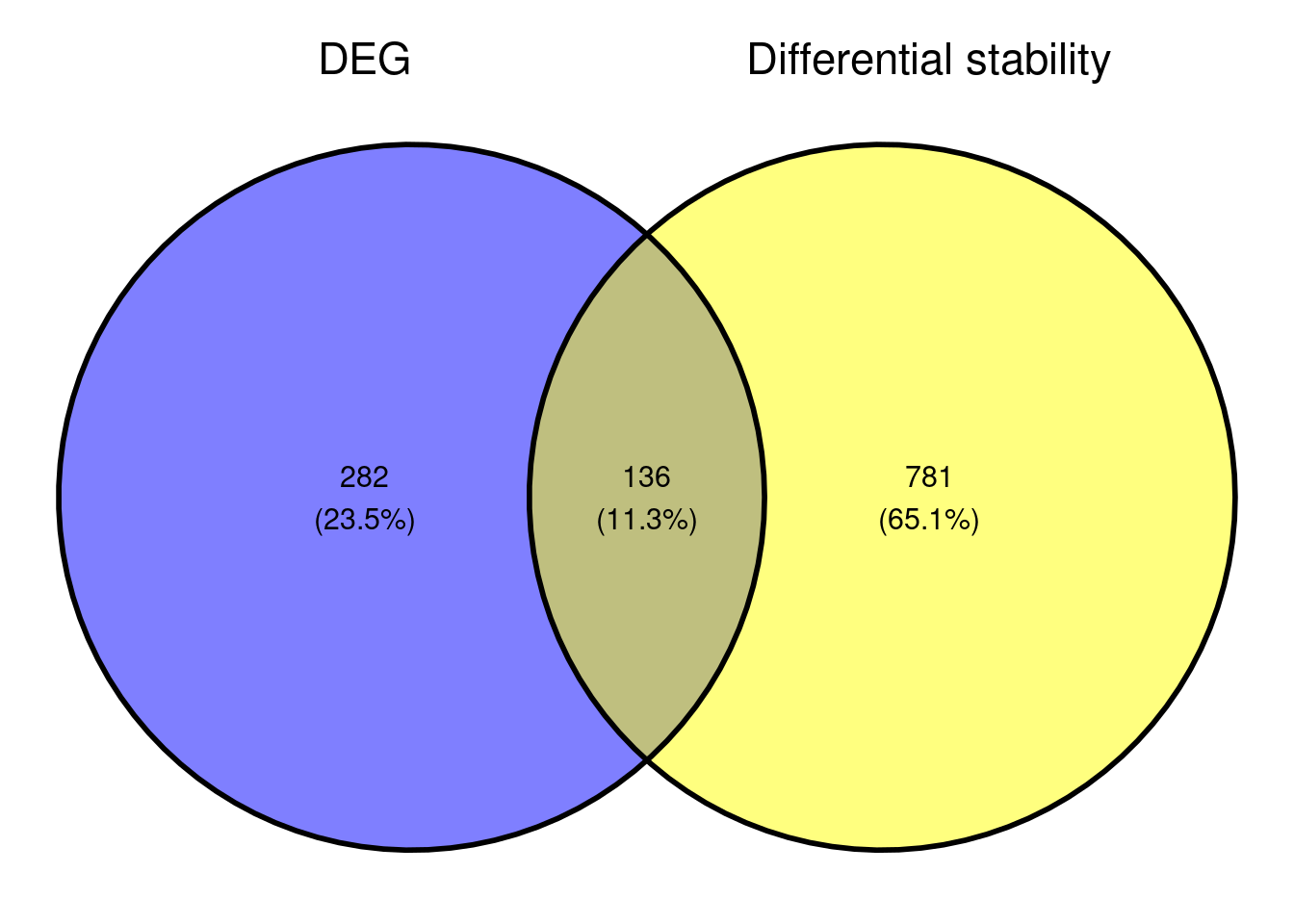
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
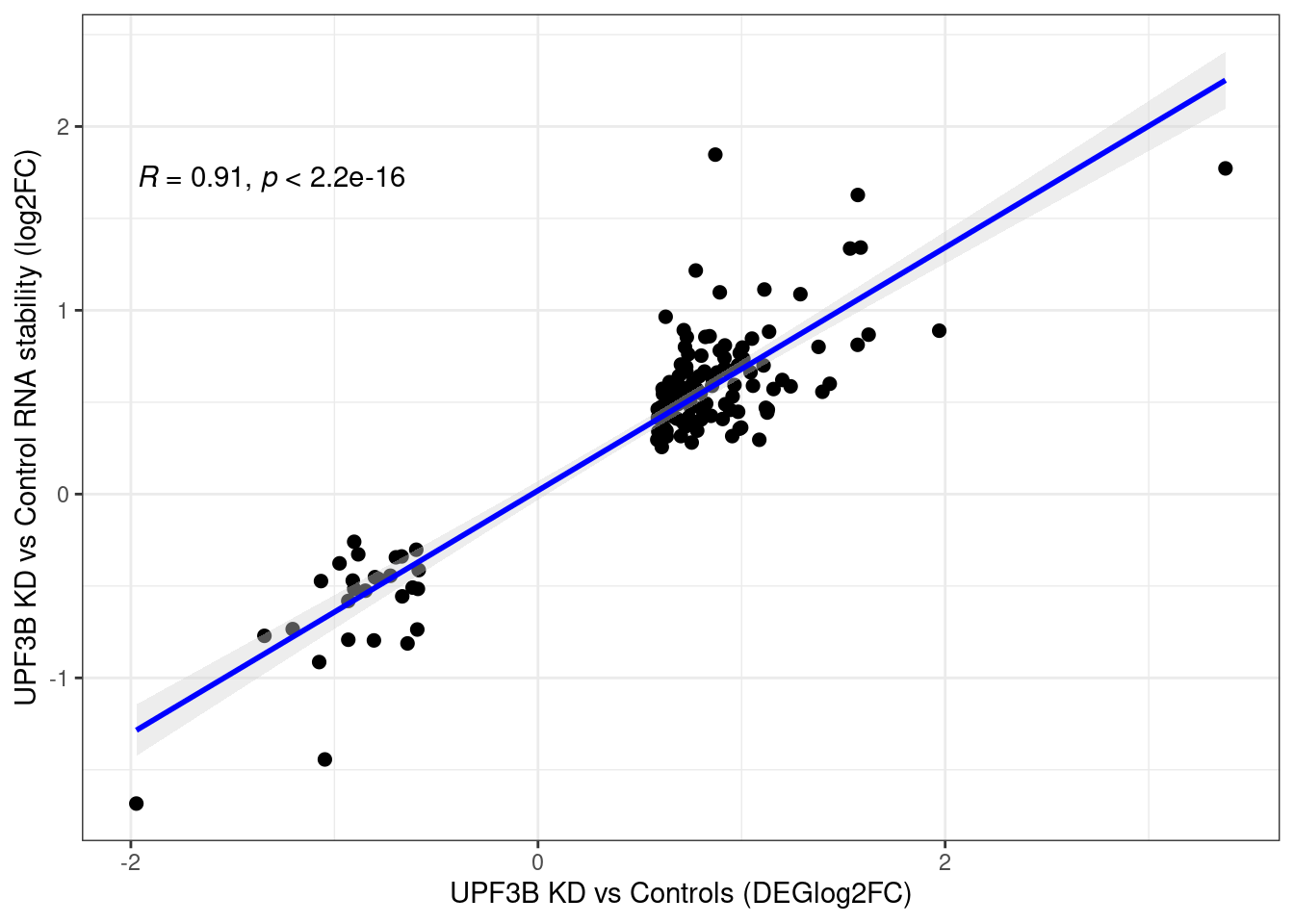
Distribution
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
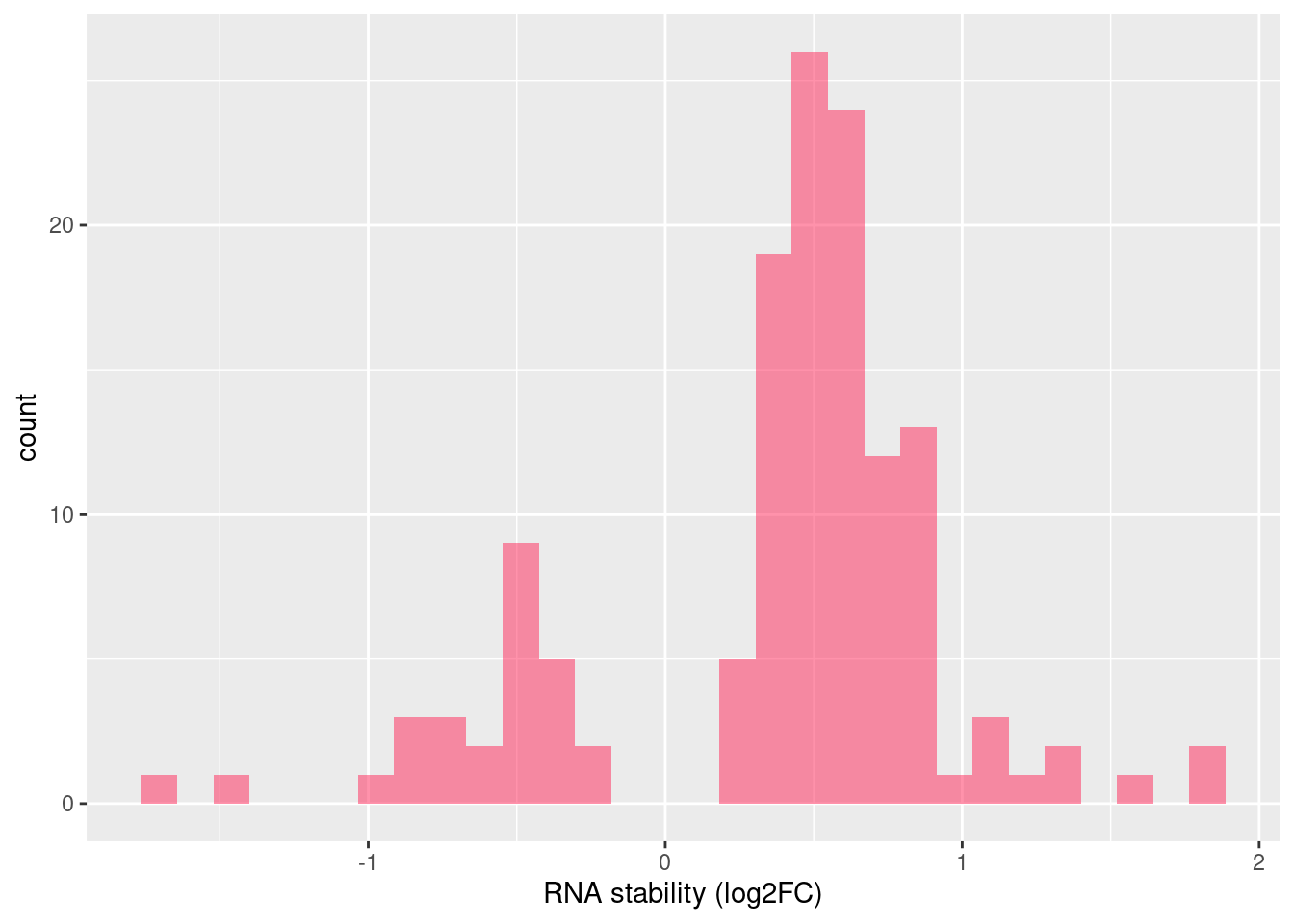
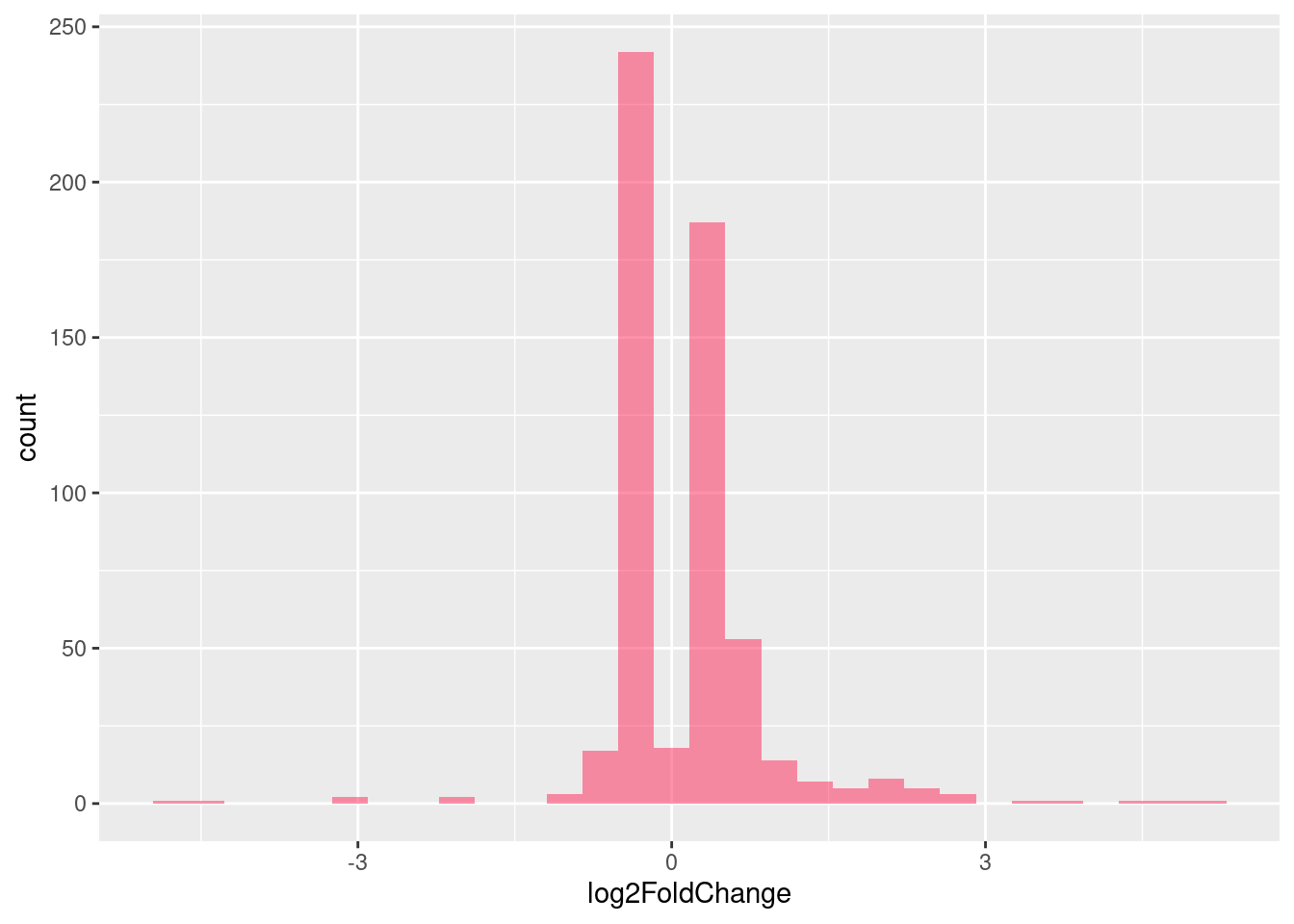
Distribution of log fold changes of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3B to Controls comparison and their distribution was plotted.
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
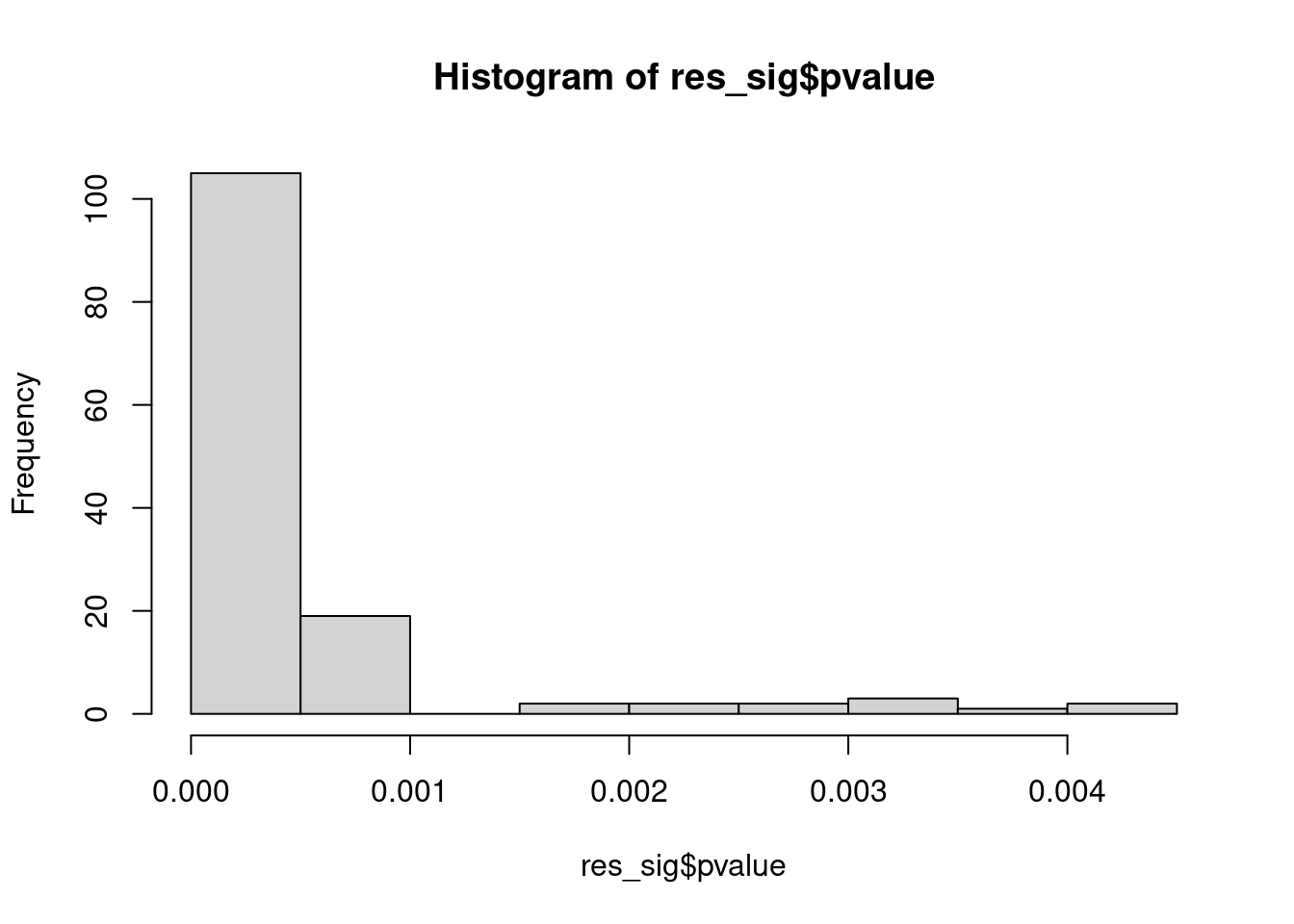
Distribution of pvalue of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3B to Controls comparison and their pvalues were plotted to ensure significance of results
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
UPF3A
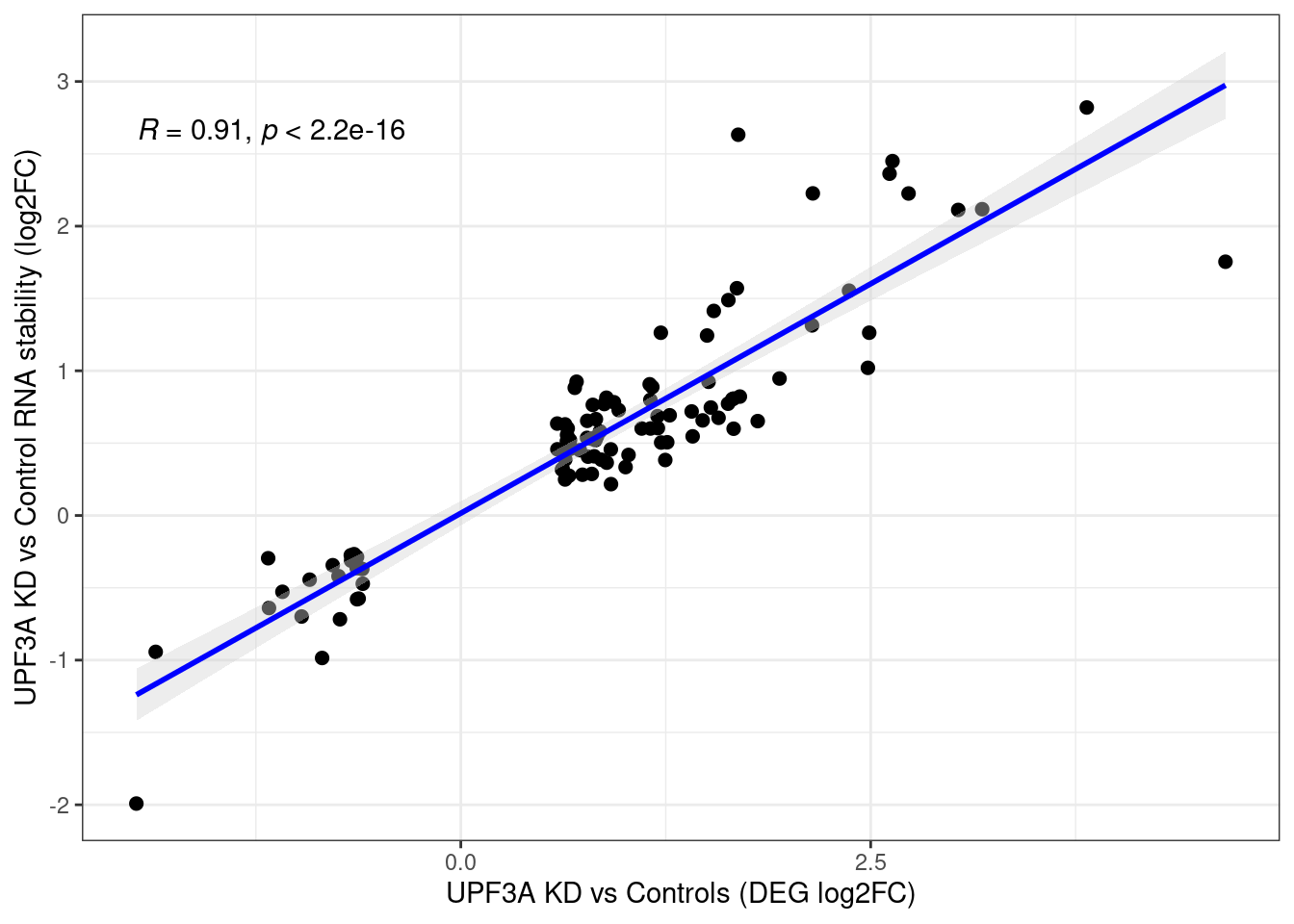
Comparison with DEGs
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
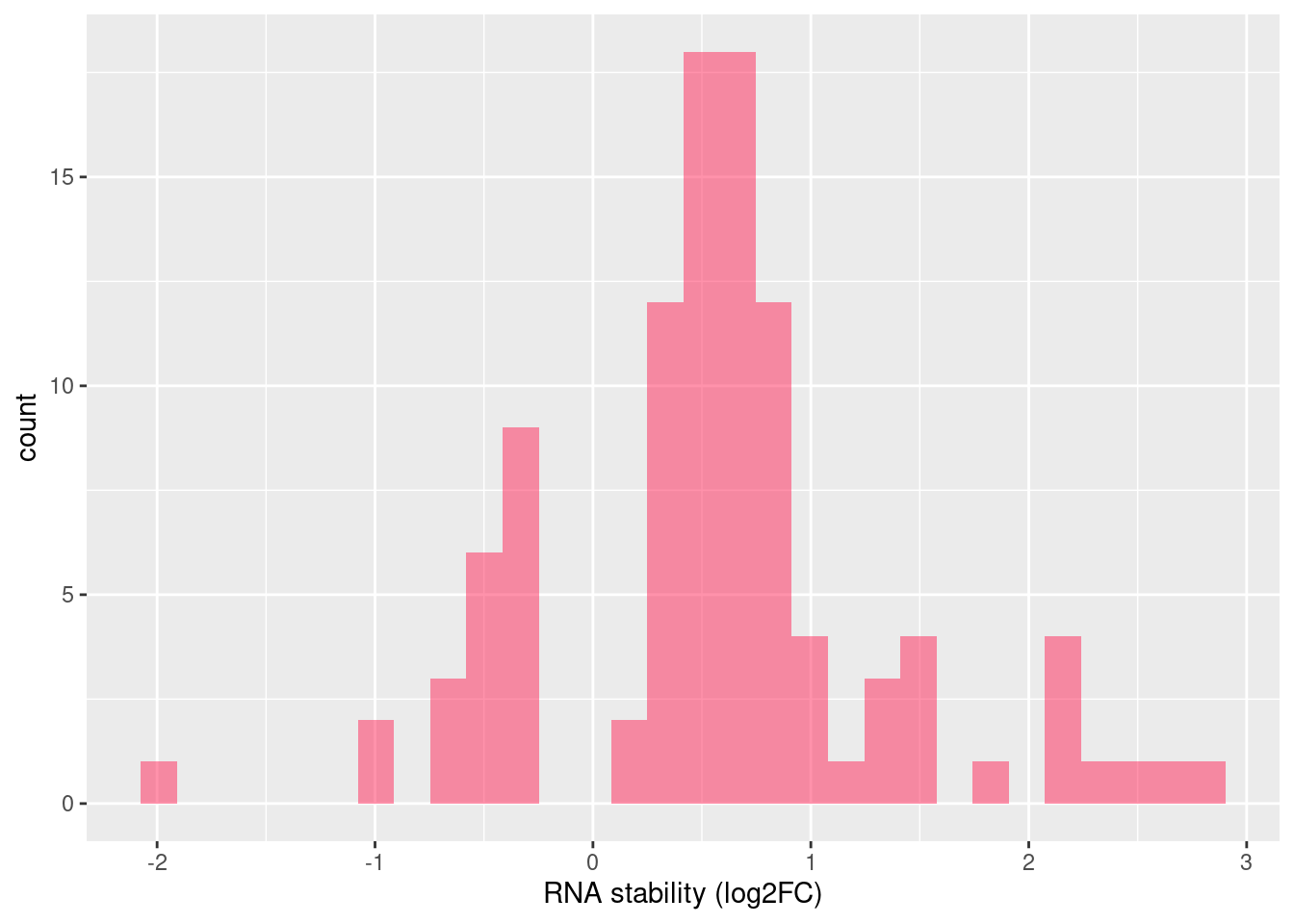
Distribution of log fold changes of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3A to Controls comparison and their distribution was plotted.
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
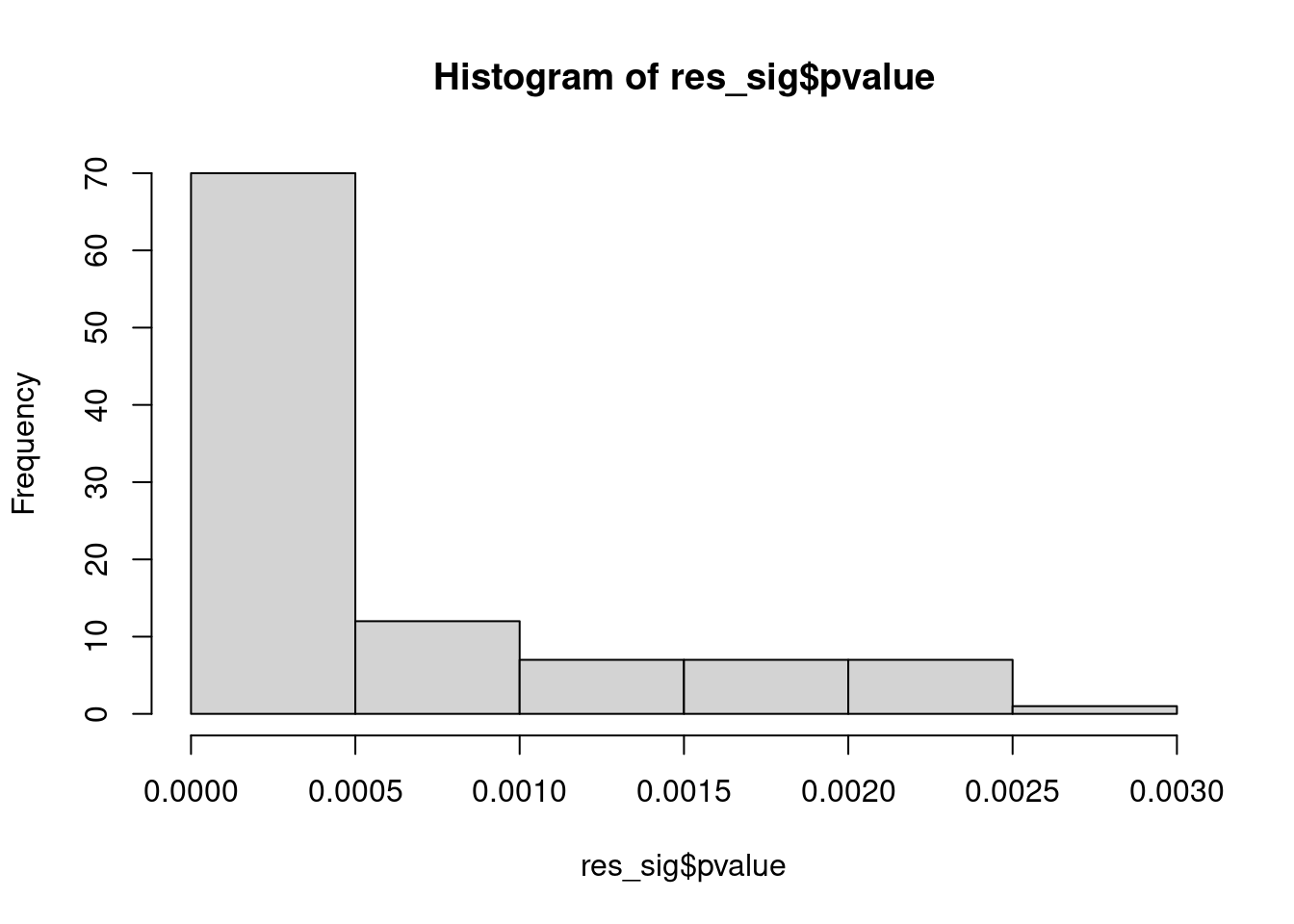
Distribution of pvalue of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3A to Controls comparison and their pvalues were plotted to ensure significance of results
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
UPF3 dKD
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
Comparison with DEGs
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
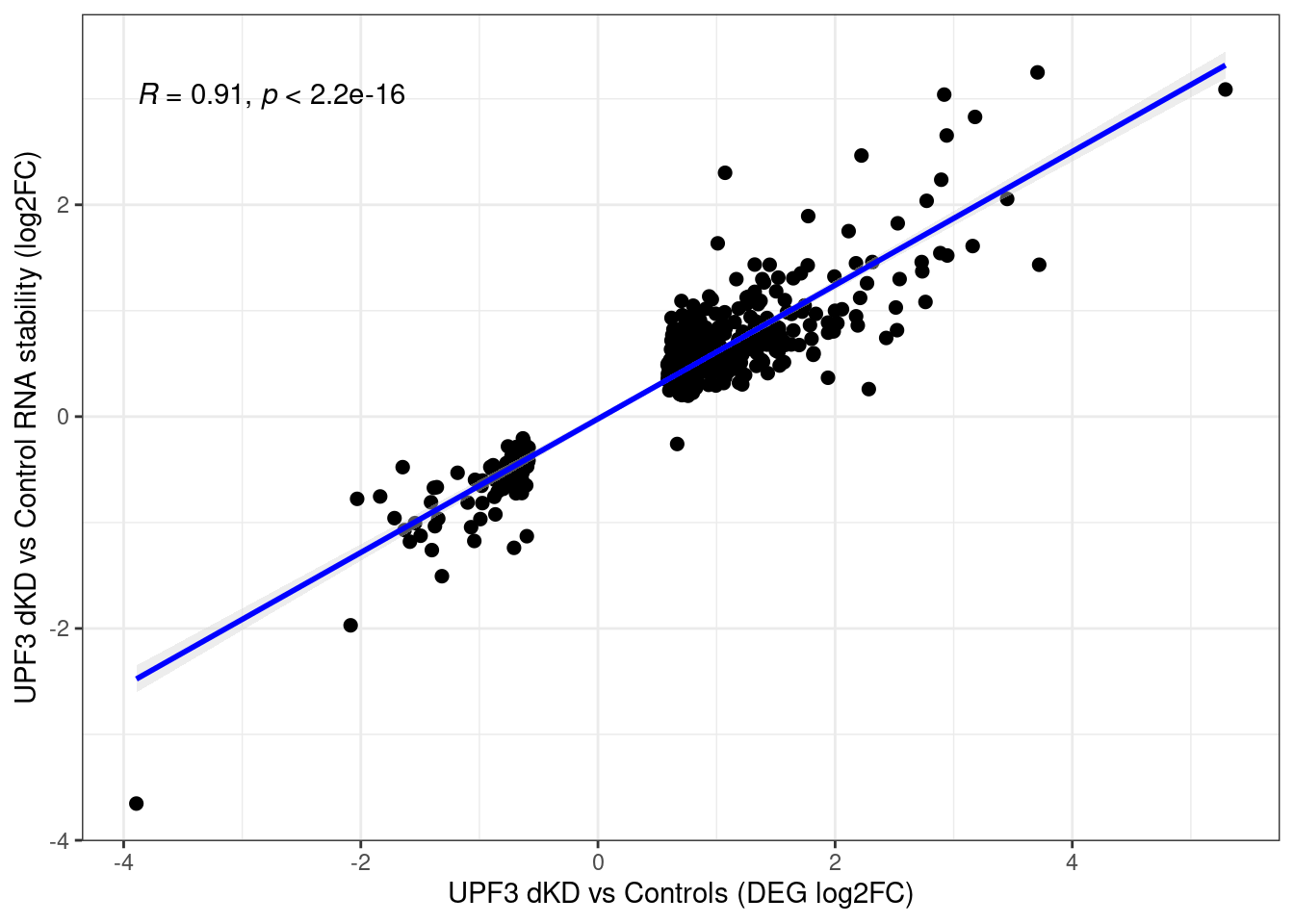
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
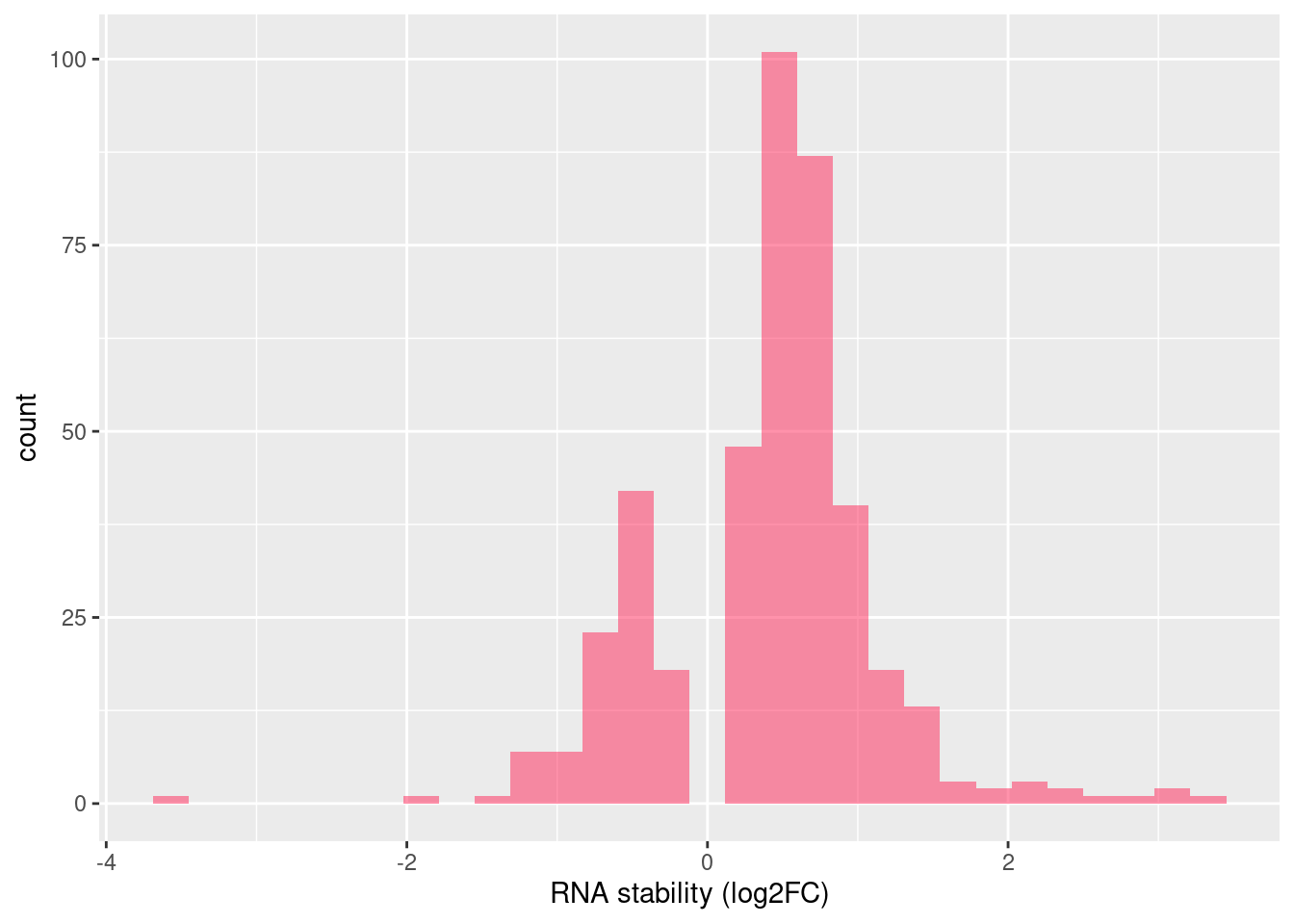
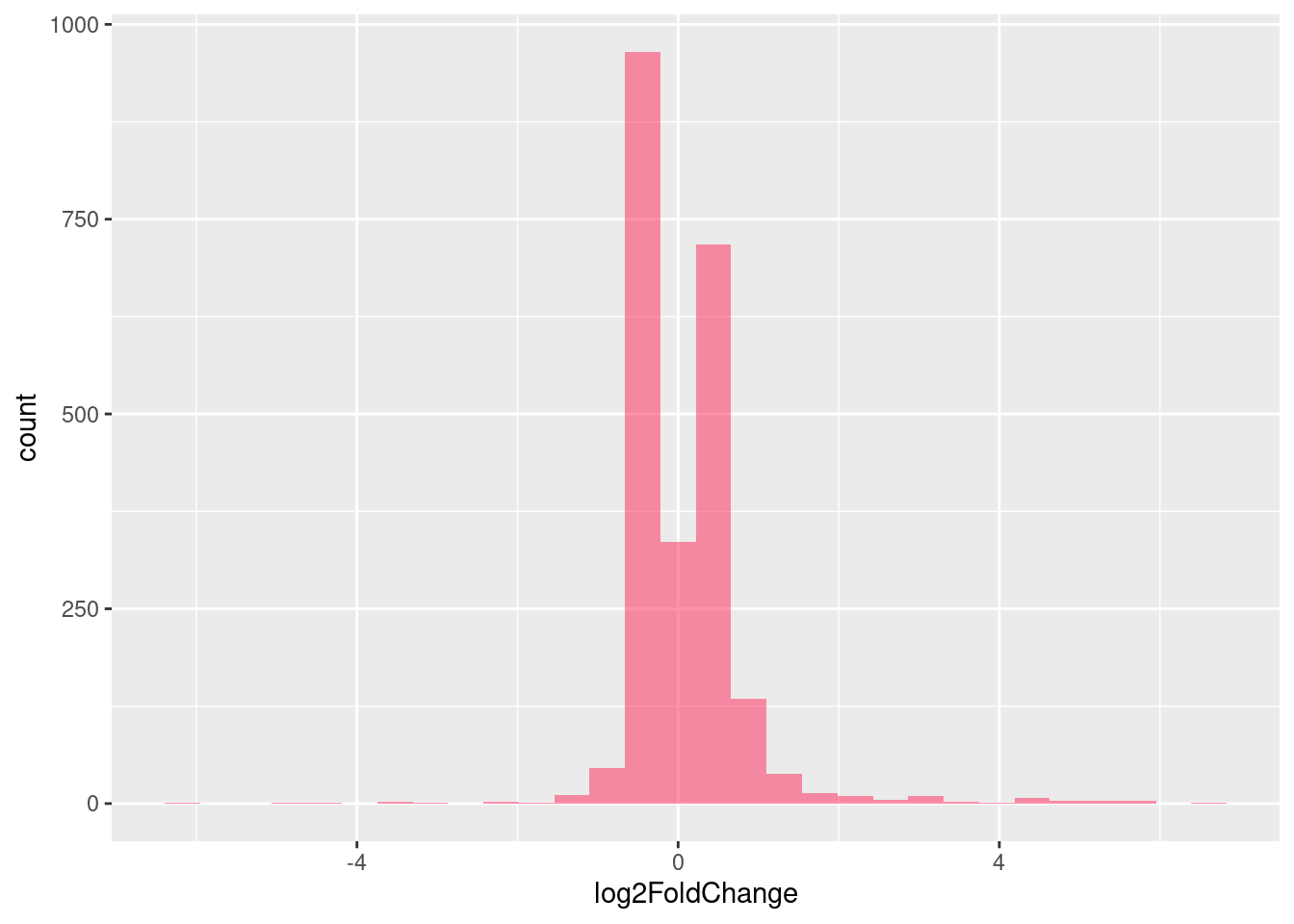
Distribution of log fold changes of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3 dKD to Controls comparison and their distribution was plotted.
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
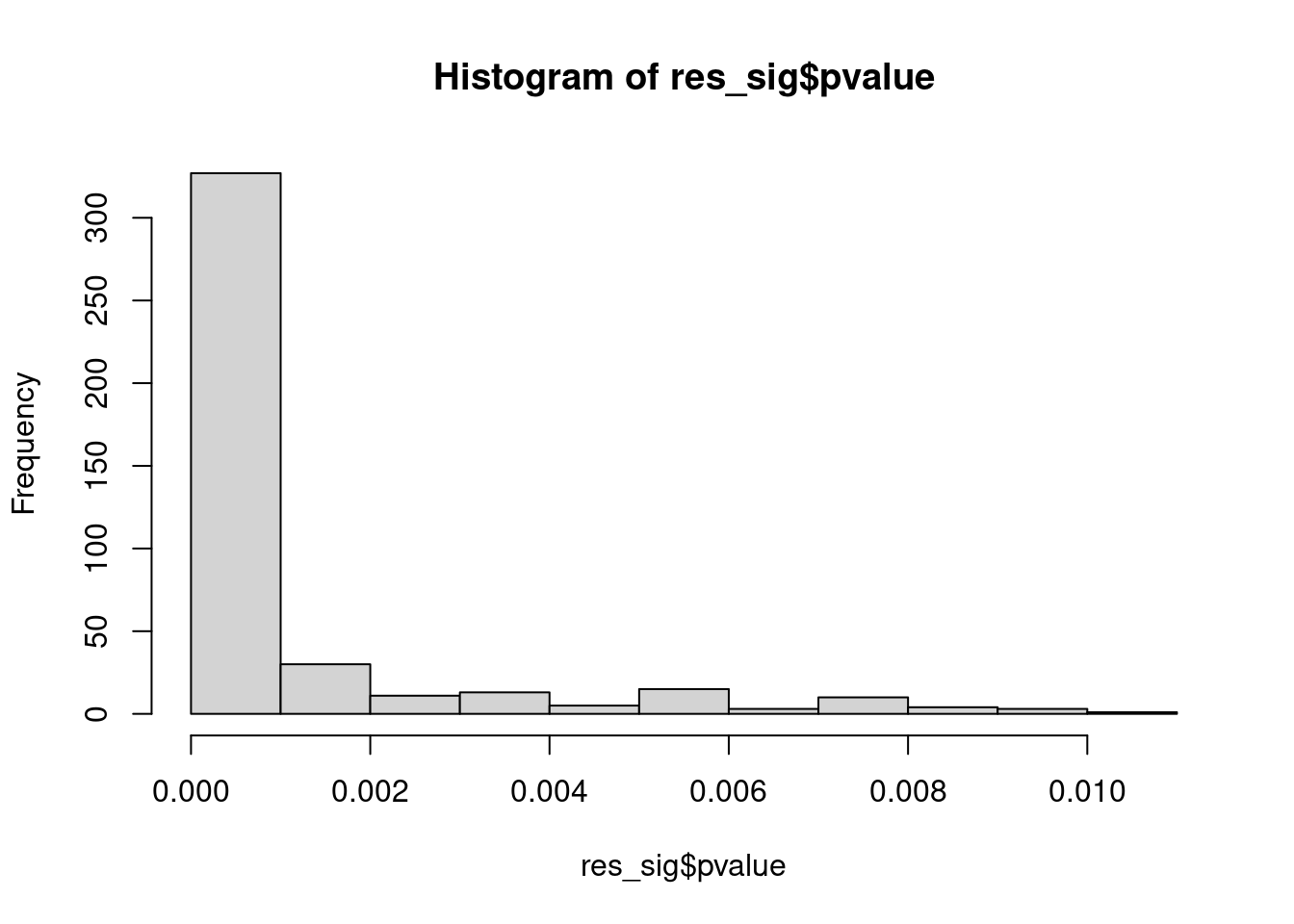
Distribution of pvalue of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3 dKD to Controls comparison and their pvalues were plotted to ensure significance of results
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
UPF3A OE UPF3B KD
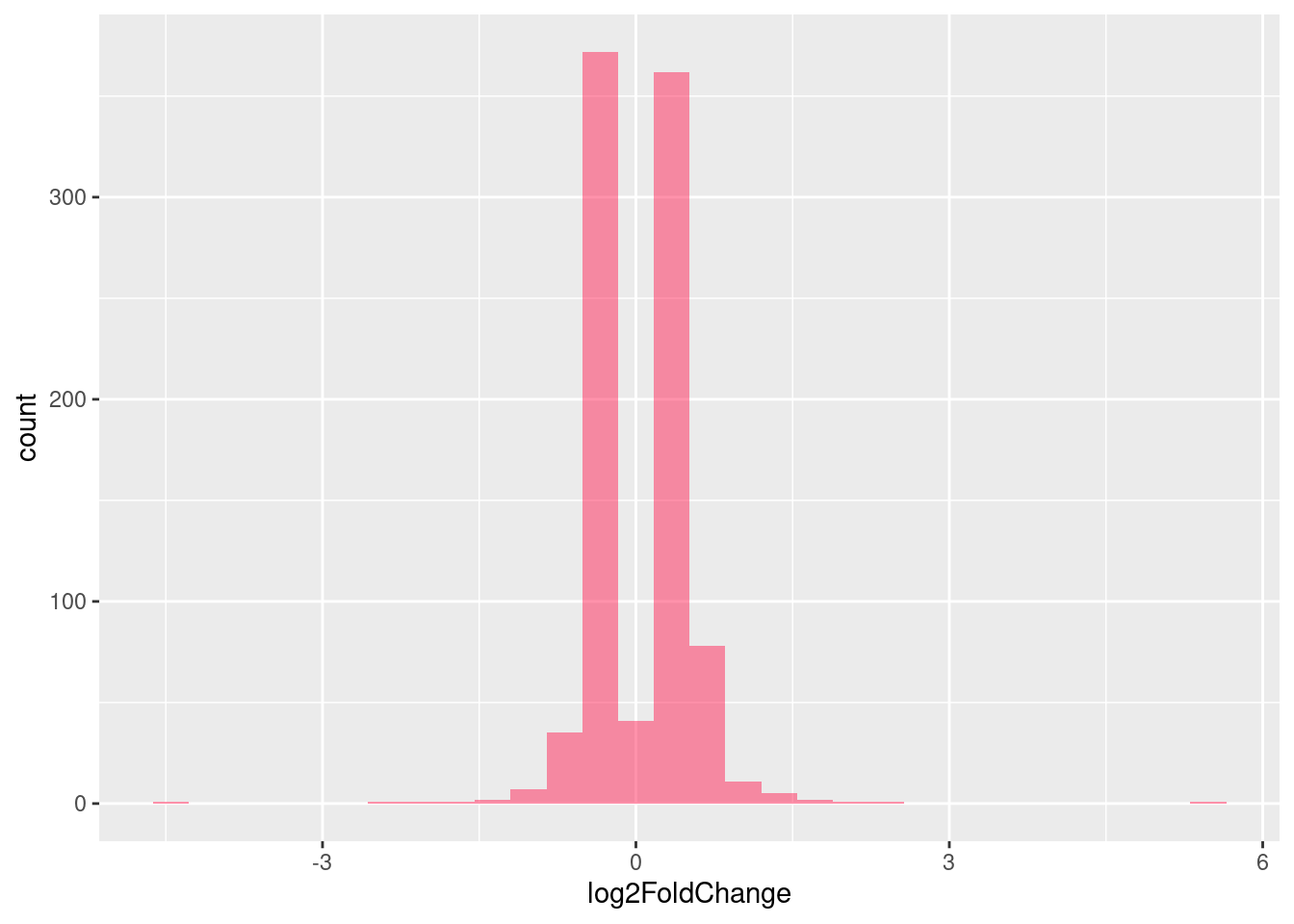
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
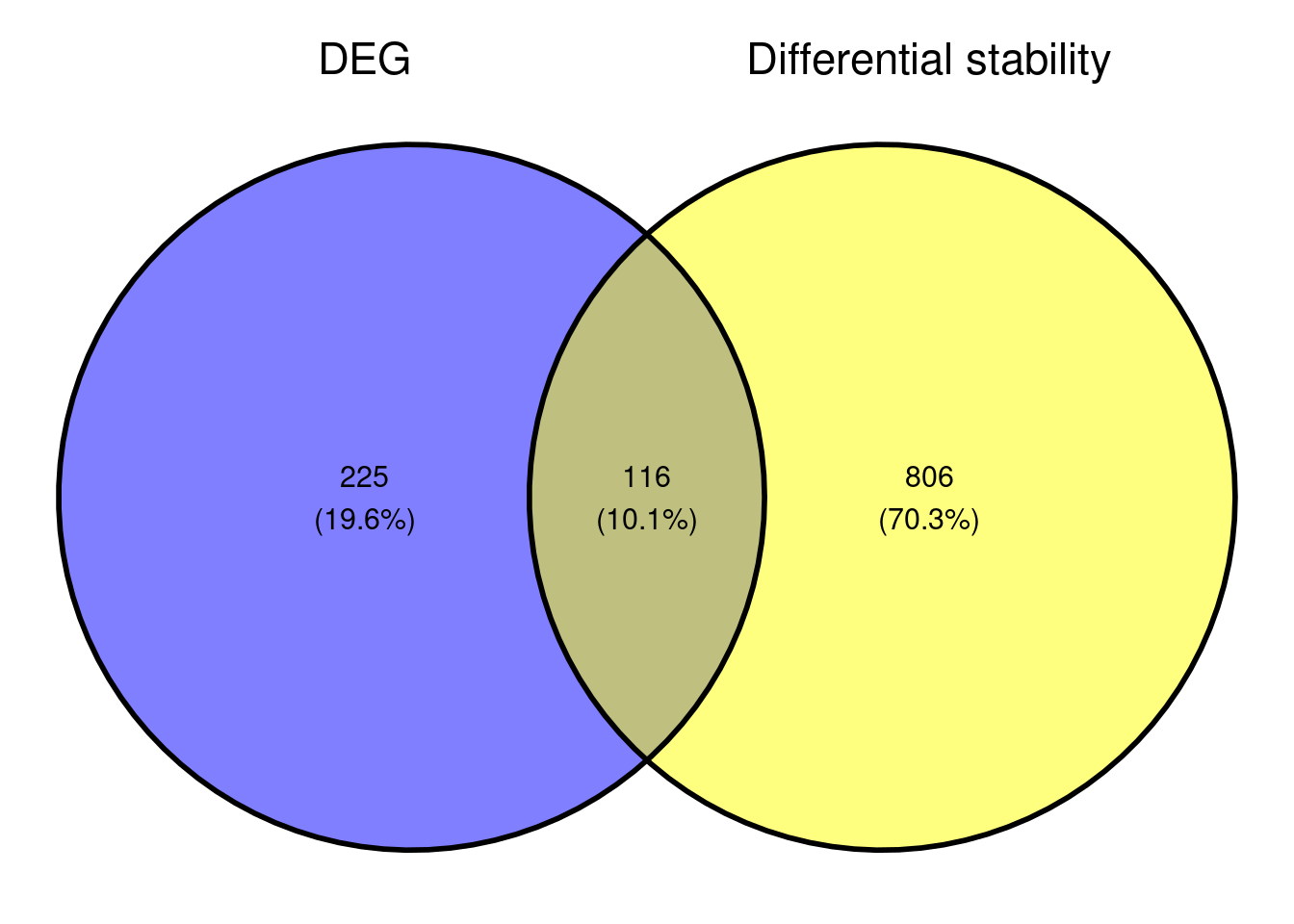
Comparison with DEGs
| Version | Author | Date |
|---|---|---|
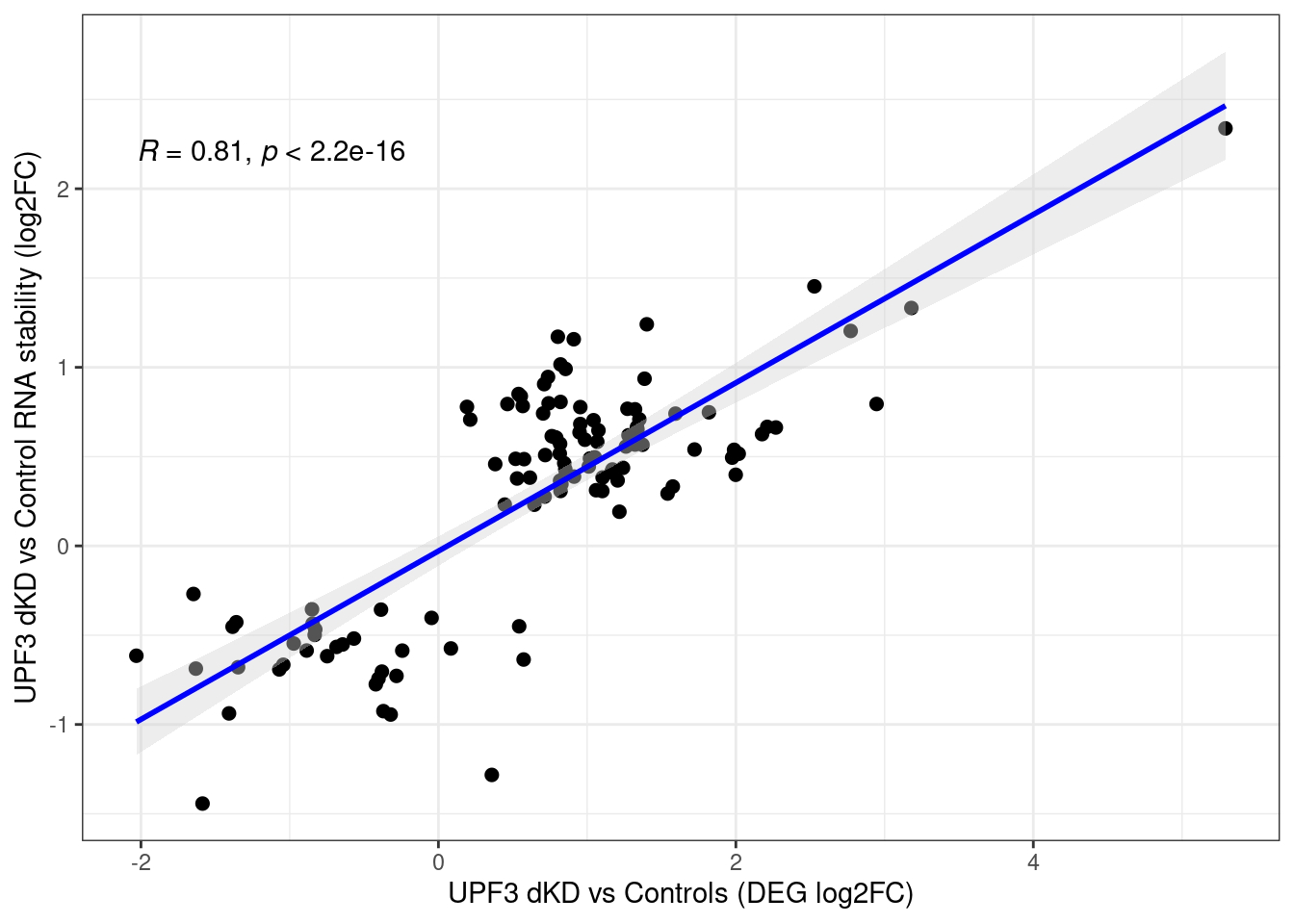
| bff9b8f | unawaz1996 | 2023-05-12 |
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
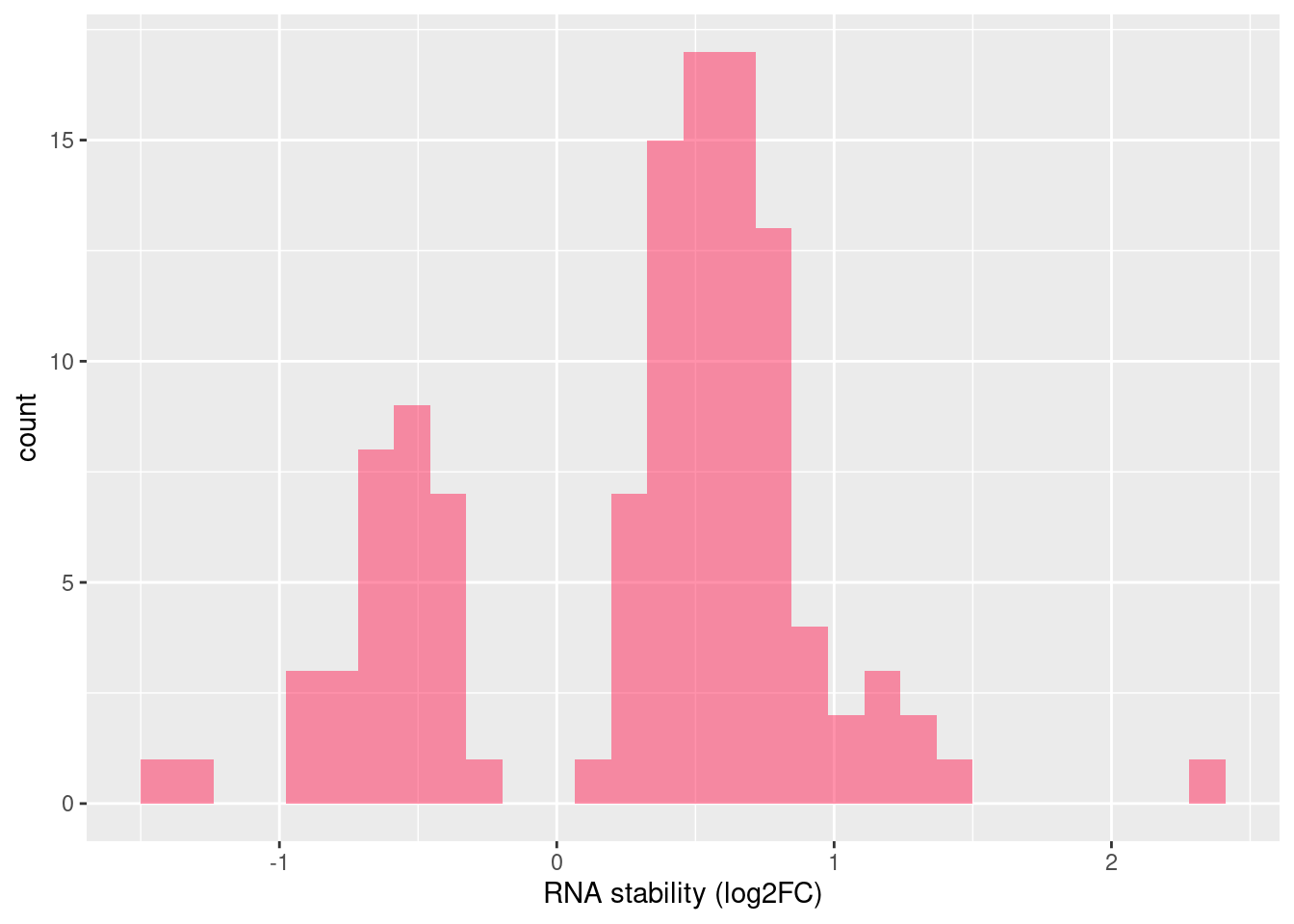
Distribution of log fold changes of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3 dKD to Controls comparison and their distribution was plotted.
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
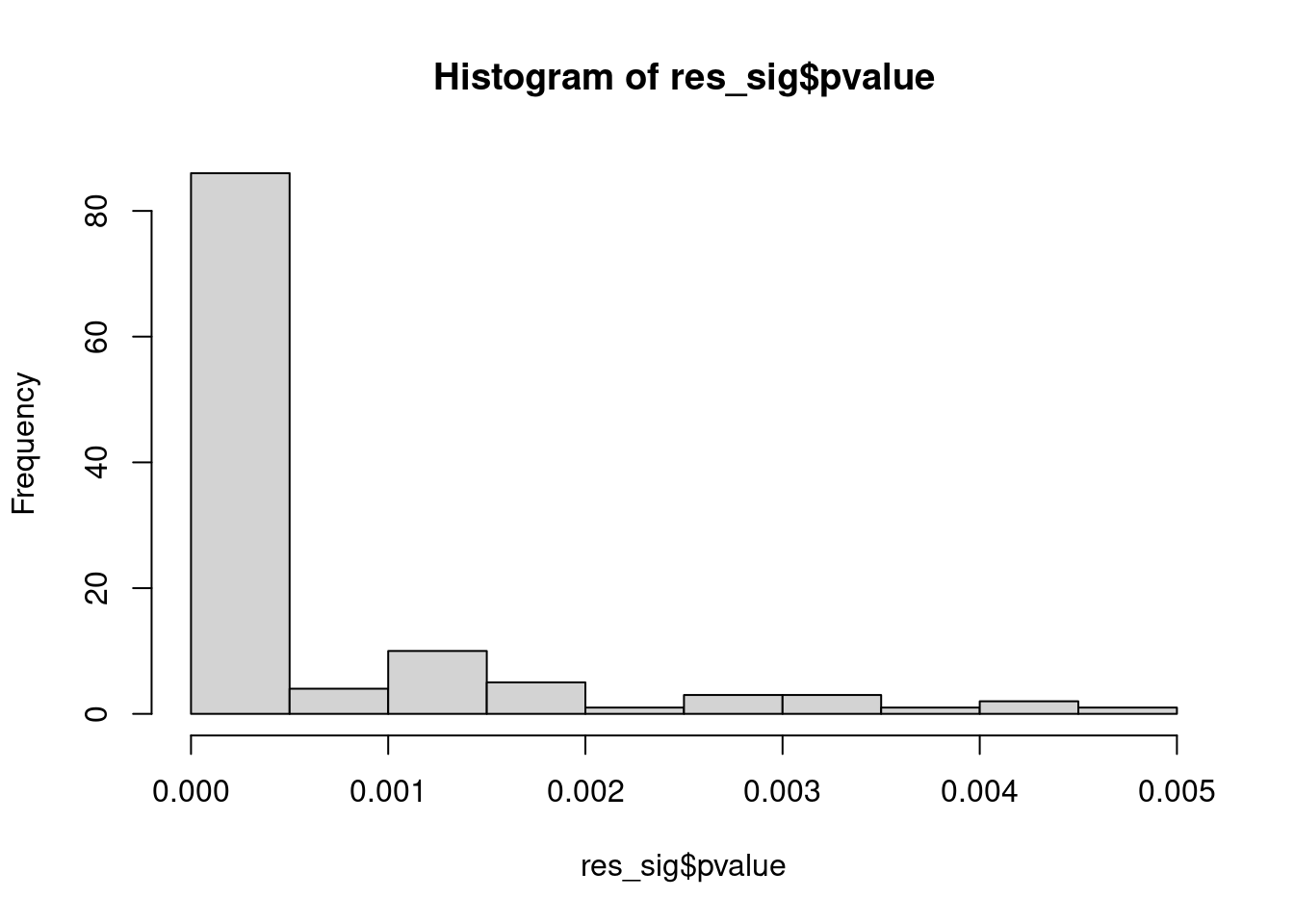
Distribution of pvalue of RNA-stability results. The genes that were significantly destabilised/stabilised were overlaapped with DEGs from UPF3 dKD to Controls comparison and their pvalues were plotted to ensure significance of results
| Version | Author | Date |
|---|---|---|
| bff9b8f | unawaz1996 | 2023-05-12 |
Gene set enrichment analysis
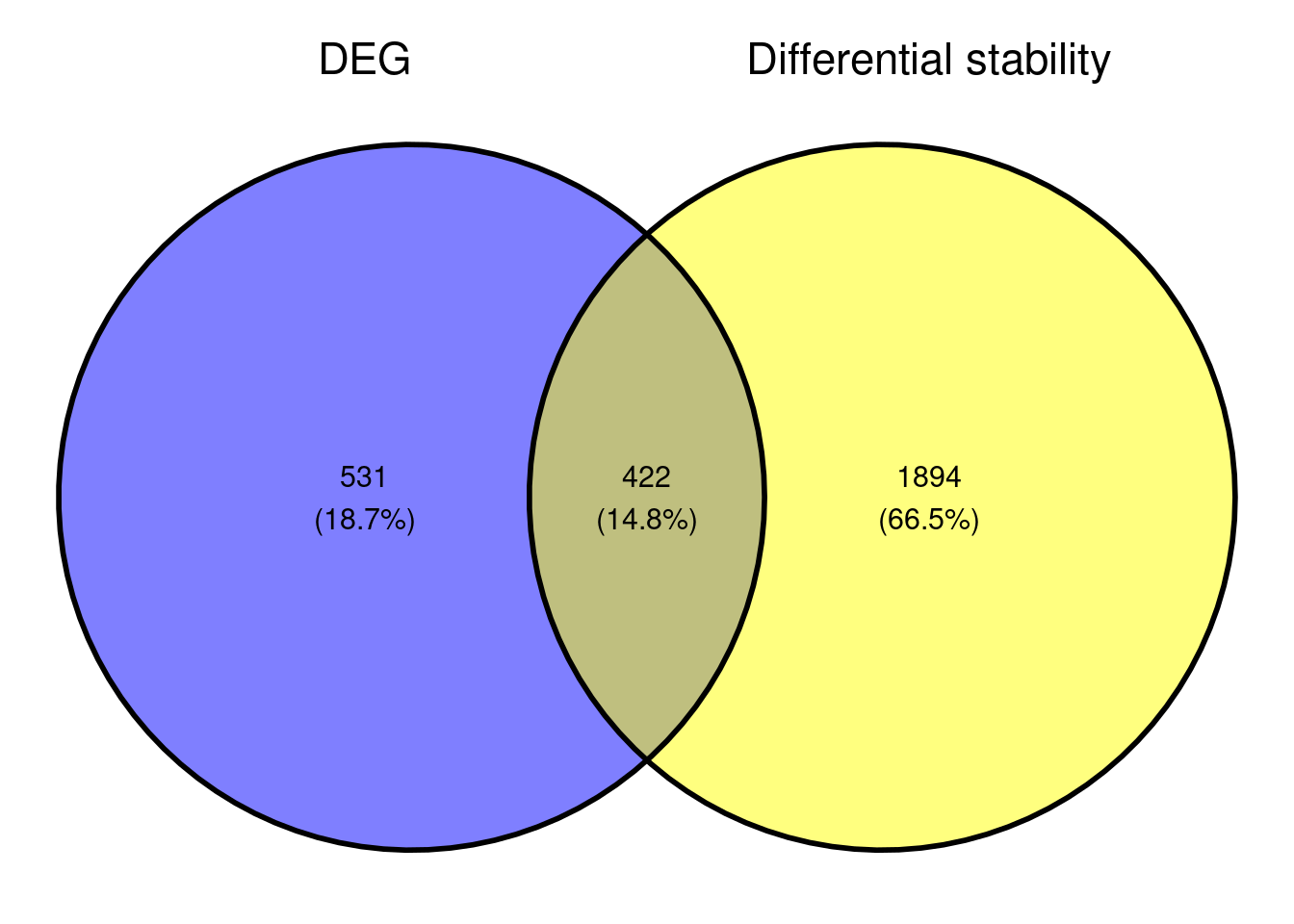
Based on the small overlap of DEGs and DSG, it might be that the genes that are differentially stabilised/destabilised are impacted as a secondary effect to NMD inhibition. It would be interesting to see what that overall data represents in terms of pathway enrichment analyses. As we are interested in seeing what gene sets are up-regulated and downregulated, we are conducting a gene set enrichment analysis.
R version 4.2.2 Patched (2022-11-10 r83330)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 tools stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] plyr_1.8.8 gplots_3.1.3
[3] coin_1.4-2 survival_3.5-5
[5] IsoformSwitchAnalyzeR_2.01.04 pfamAnalyzeR_0.99.0
[7] sva_3.46.0 genefilter_1.80.3
[9] mgcv_1.8-42 nlme_3.1-162
[11] satuRn_1.6.0 DEXSeq_1.44.0
[13] BiocParallel_1.32.6 ggrepel_0.9.3
[15] pander_0.6.5 msigdbr_7.5.1
[17] cowplot_1.1.1 ngsReports_2.0.3
[19] patchwork_1.1.2 VennDiagram_1.7.3
[21] futile.logger_1.4.3 UpSetR_1.4.0
[23] fgsea_1.24.0 GOplot_1.0.2
[25] RColorBrewer_1.1-3 gridExtra_2.3
[27] ggdendro_0.1.23 AnnotationHub_3.6.0
[29] BiocFileCache_2.6.1 dbplyr_2.3.2
[31] openxlsx_4.2.5.2 ggiraph_0.8.7
[33] wasabi_1.0.1 sleuth_0.30.1
[35] DT_0.27 VennDetail_1.14.0
[37] msigdb_1.6.0 GSEABase_1.60.0
[39] graph_1.76.0 annotate_1.76.0
[41] XML_3.99-0.14 pheatmap_1.0.12
[43] ggvenn_0.1.10 MetBrewer_0.2.0
[45] ggpubr_0.6.0 venn_1.11
[47] viridis_0.6.2 viridisLite_0.4.1
[49] tximeta_1.16.1 tximport_1.26.1
[51] goseq_1.50.0 geneLenDataBase_1.34.0
[53] BiasedUrn_2.0.9 org.Mm.eg.db_3.16.0
[55] EnsDb.Mmusculus.v79_2.99.0 ensembldb_2.22.0
[57] AnnotationFilter_1.22.0 GenomicFeatures_1.50.4
[59] AnnotationDbi_1.60.2 biomaRt_2.54.1
[61] edgeR_3.40.2 limma_3.54.2
[63] DESeq2_1.38.3 SummarizedExperiment_1.28.0
[65] Biobase_2.58.0 MatrixGenerics_1.10.0
[67] matrixStats_0.63.0 GenomicRanges_1.50.2
[69] GenomeInfoDb_1.34.9 IRanges_2.32.0
[71] S4Vectors_0.36.2 BiocGenerics_0.44.0
[73] corrplot_0.92 lubridate_1.9.2
[75] forcats_1.0.0 purrr_1.0.1
[77] readr_2.1.4 tidyverse_2.0.0
[79] stringr_1.5.0 tidyr_1.3.0
[81] scales_1.2.1 data.table_1.14.8
[83] readxl_1.4.2 tibble_3.2.1
[85] magrittr_2.0.3 reshape2_1.4.4
[87] ggplot2_3.4.2 dplyr_1.1.1
[89] eisaR_1.10.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 rtracklayer_1.58.0
[3] bit64_4.0.5 knitr_1.42
[5] multcomp_1.4-23 DelayedArray_0.24.0
[7] hwriter_1.3.2.1 KEGGREST_1.38.0
[9] RCurl_1.98-1.12 generics_0.1.3
[11] TH.data_1.1-1 callr_3.7.3
[13] lambda.r_1.2.4 RSQLite_2.3.1
[15] bit_4.0.5 tzdb_0.3.0
[17] xml2_1.3.3 httpuv_1.6.9
[19] xfun_0.38 hms_1.1.3
[21] jquerylib_0.1.4 babelgene_22.9
[23] evaluate_0.20 promises_1.2.0.1
[25] fansi_1.0.4 restfulr_0.0.15
[27] progress_1.2.2 caTools_1.18.2
[29] DBI_1.1.3 geneplotter_1.76.0
[31] htmlwidgets_1.6.2 ellipsis_0.3.2
[33] crosstalk_1.2.0 backports_1.4.1
[35] libcoin_1.0-9 locfdr_1.1-8
[37] vctrs_0.6.1 abind_1.4-5
[39] cachem_1.0.7 withr_2.5.0
[41] BSgenome_1.66.3 GenomicAlignments_1.34.1
[43] prettyunits_1.1.1 lazyeval_0.2.2
[45] crayon_1.5.2 labeling_0.4.2
[47] pkgconfig_2.0.3 ProtGenerics_1.30.0
[49] rlang_1.1.0 lifecycle_1.0.3
[51] sandwich_3.0-2 filelock_1.0.2
[53] cellranger_1.1.0 rprojroot_2.0.3
[55] Matrix_1.5-3 carData_3.0-5
[57] boot_1.3-28.1 Rhdf5lib_1.20.0
[59] zoo_1.8-11 whisker_0.4.1
[61] processx_3.8.0 png_0.1-8
[63] rjson_0.2.21 bitops_1.0-7
[65] getPass_0.2-2 KernSmooth_2.23-20
[67] rhdf5filters_1.10.1 Biostrings_2.66.0
[69] blob_1.2.4 rstatix_0.7.2
[71] ggsignif_0.6.4 memoise_2.0.1
[73] zlibbioc_1.44.0 compiler_4.2.2
[75] BiocIO_1.8.0 Rsamtools_2.14.0
[77] cli_3.6.1 XVector_0.38.0
[79] pbapply_1.7-0 ps_1.7.4
[81] formatR_1.14 MASS_7.3-58.3
[83] tidyselect_1.2.0 stringi_1.7.12
[85] highr_0.10 yaml_2.3.7
[87] locfit_1.5-9.7 sass_0.4.5
[89] fastmatch_1.1-3 timechange_0.2.0
[91] parallel_4.2.2 rstudioapi_0.14
[93] uuid_1.1-0 git2r_0.31.0
[95] farver_2.1.1 digest_0.6.31
[97] BiocManager_1.30.20 shiny_1.7.4
[99] Rcpp_1.0.10 car_3.1-2
[101] broom_1.0.4 BiocVersion_3.16.0
[103] later_1.3.0 httr_1.4.5
[105] colorspace_2.1-0 fs_1.6.1
[107] splines_4.2.2 statmod_1.5.0
[109] plotly_4.10.1 systemfonts_1.0.4
[111] xtable_1.8-4 jsonlite_1.8.4
[113] futile.options_1.0.1 modeltools_0.2-23
[115] R6_2.5.1 pillar_1.9.0
[117] htmltools_0.5.5 mime_0.12
[119] glue_1.6.2 fastmap_1.1.1
[121] interactiveDisplayBase_1.36.0 codetools_0.2-19
[123] mvtnorm_1.1-3 utf8_1.2.3
[125] lattice_0.20-45 bslib_0.4.2
[127] curl_5.0.0 gtools_3.9.4
[129] zip_2.2.2 GO.db_3.16.0
[131] admisc_0.31 rmarkdown_2.21
[133] munsell_0.5.0 rhdf5_2.42.0
[135] GenomeInfoDbData_1.2.9 gtable_0.3.3